
Nutritionally deficient EED chips recapitulate EED patient transcriptional signatures
We previously described a two-channel microfluidic human intestine chip, lined with living human intestinal epithelium isolated from patient-derived organoids, that undergoes villus differentiation, accumulates mucus and exhibits many features of living human intestine when cultured on-chip under continuous flow with peristalsis-like mechanical deformations28 (Fig. 1a). Additionally, transcriptional analysis demonstrated that when lined by organoid-derived duodenal epithelium, this intestine chip more closely mimicked in vivo human duodenum than the organoids used to create the chips28. To define the contribution of the intestinal epithelium to the EED phenotype, we created intestine chips lined with intestinal epithelial cells from organoids derived from surgical biopsies of either healthy or EED patient duodenum (healthy chips and EED chips, respectively). Compared with healthy chips, EED chips showed differential expression of 287 genes (q < 0.05 and fold change ≥1.5, q = FDR-adjusted-p-value; 86 upregulated, 201 downregulated) (Fig. 1b). EED chips showed upregulation of MUC5AC (previously shown to reduce inflammation, intestinal injury and bacterial translocation in an experimental intestinal injury35), neuregulin-4 (a known survival factor for colonic epithelium that protects against experimental intestinal injury36,37) and the intestinal stem cell marker SMOC2, whereas brush border peptidase MME, oxidative stress and inflammatory response controlling ectoenzyme VNN1, tight junction protein CLDN10 and secreted goblet cell protein CLCA1 were all downregulated (Supplementary Table 1).
a, A schematic representation of small-intestine chips. b, Compared with healthy chips, the EED transcriptome consists of 287 differentially expressed genes (red; q < 0.05 and fold change ≥1.5). Exposure of healthy and EED chips to -N/-T media resulted in an increased number of differentially expressed genes with 690 (yellow) and 969 genes (blue), respectively. Of these, 307 genes were differentially expressed in both healthy and EED chips exposed to nutritional deficiency (yellow and blue). Seventy-one genes were differentially expressed in EED chips when compared with healthy chips in control medium that are further affected by the addition of nutritionally deficient medium (blue and red, respectively; 3 chips for each condition). Each chip corresponds to one biological replicate. c, A comparison of the 20 most up- and 20 most downregulated genes from the clinical EED signature, with healthy or EED chip gene expression depicted as a heat map (red, upregulation; blue, downregulation) showing that EED -N/-T has the closest hierarchical relationship to the clinical EED signature (clinical EED Z-score distance from intestine chips results was 15.2 for the EED -N/-T vs EED; 18.7 for the healthy -N/-T vs healthy control; and 19.4 for EED control vs healthy control). The comparison for EED -N/-T to healthy control is similar and shown in Extended Data Fig. 1. Ten dendrogram gene clusters were also defined and the corresponding roots enumerated; 3 chips for each condition. Each chip corresponds to one biological replicate. d, Of the top 9 upregulated genes in the clinical EED signature, 6 were also upregulated when EED chips were exposed to -N/-T media; 3 chips for each condition. Each chip corresponds to one biological replicate. e, Functional pathway analysis was performed using the contextual language-processing programme COMPBIO. The spatial map depicts themes related to differentially expressed genes as nodes, with interconnections depicted as edges whose thickness relates to the degree of interconnectedness. The themes were also ranked according to a score representing fold enrichment over random clustering and this score determines sphere size (Supplementary Table 2); 3 chips for each condition. Each chip corresponds to one biological replicate.
We compared this differential gene expression profile with a recently derived clinical EED signature, which was obtained by comparing profiles of intestinal tissue samples from EED patients who were also refractory to nutritional intervention (Study of Environmental Enteropathy and Malnutrition, SEEM)38 versus samples from healthy control patients who were investigated for gastrointestinal symptoms but had normal endoscopic and histologic findings (Cincinnati Children’s Hospital Medical Center). When we compared gene profiles from EED chips versus healthy chips cultured in control medium (that is, with all nutrients present), the differentially expressed genes had some overlap with the clinical EED signature, including most notably a shared downregulation of metallothioneins (MT1X, MT1A, MT1F, MT1H, MSMB and MT1M; gene dendrogram cluster 5) (Fig. 1c).
We then carried out the same experiment but perfused both the healthy and EED intestine chips with medium deficient in niacinamide and tryptophan (−N/−T), selected on the basis of past work implicating their role in EED15,16,19. When we compared expression profiles from the healthy chips exposed to nutritional deficiency (healthy −N/−T chip) versus the healthy control chips, we detected differential expression of 690 genes (q < 0.05 and fold change ≥1.5; 556 upregulated, 124 downregulated) (Fig. 1b and Supplementary Fig. 1), including upregulation of the amino acid starvation-related transcription factor ATF4, its downstream solute carriers (SLC34A2, SLC7A5 and SLC6A9) and the inflammation-associated gene LCN2 (Supplementary Table 1). Uniformity of transcriptome analysis of samples in each group was confirmed by principal component analysis (Supplementary Fig. 2). There also appeared to be a trend towards upregulation of several antimicrobial and immune response genes as seen in the clinical EED signature, but these changes did not reach statistical significance. While there was greater overlap between the transcriptome of the healthy −N/−T chip with the clinical EED transcriptome than observed with the EED control chip compared with healthy control chip, some genes were regulated in an opposing direction, including upregulation of metallothioneins (Fig. 1c, gene dendrogram cluster 5).
In contrast, we observed closer unsupervised hierarchical clustering with the clinical EED signature when the EED chip was exposed to nutritional deficiency (EED −N/−T chip vs EED control chip) (Fig. 1c). Culture of the EED intestine chips in −N/−T medium yielded differential expression of 969 genes (q < 0.05 and fold change ≥1.5; 522 upregulated, 447 downregulated) (Fig. 1b). This was manifested by the upregulation of antimicrobial genes (SAA1, SAA2, DUOXA2, DUOX2 and CXCL5; gene dendrogram clusters 1, 3 and 4) and downregulation of not only metallothioneins but also metabolic and digestive genes (SLC6A14 and GUC2AB; gene dendrogram clusters 4 and 9). This congruence was most noticeable among the top 10 upregulated genes of the clinical EED signature, 6 of which (60%) were also upregulated when EED chips were exposed to nutritionally deficient medium (Fig. 1d).
To identify pathways affected by exposure of EED intestine chips to −N/−T nutritional deficiency, we used a contextual language-processing programme to identify and rank functionally related clusters of genes39. This analysis revealed several pathways that were significantly upregulated when EED chips were exposed to −N/−T media, including a chemokine pathway (score 1,833.89, indicates fold enrichment over random association) and a pathway associated with amino acid starvation (score 1,988.09) (Fig. 1e and Supplementary Table 2). Within the amino acid starvation pathway, the ATF4 gene is upstream of several other pathways including tRNA metabolism (score 1,547.01), DNA damage (score 1,790.28), p53 methylation (score 1,608.44) and amino acid transporters (score 2,022.97). Many of these same genes were also upregulated in nutritionally deficient healthy −N/−T chips, including ATF4, TP53, AARS, YARS, MDM2, CCND2 and several amino acid transporters (Supplementary Table 1). Conversely, nutritional deficiency led to the downregulation of pathways related to fatty acid uptake (score 3,590.64), brush border structural integrity (score 3,777.69), mitosis (score 3,311.49), cytochrome p450 (score 2,581.54) and fatty acid utilization (score 2,056.86) in the EED chips. These results are consistent with the observation that the intestines of nutritionally deficient EED patients are characterized by having decreased brush border development and impaired cell growth40,41,42.
We also compared gene expression changes in our model with previously identified clusters of intestinal cell-type gene markers43,44,45. In control medium, enterocyte markers were expressed at lower levels in EED chips compared with healthy chips (in particular, MME, APOB and MTTP were reduced by 15-fold, 2.7-fold and 2.3-fold, respectively) and were even more broadly downregulated in EED chips that were exposed to nutritional deficiency (in particular, PCK1, MEP1B and CREB3L3 were decreased by 7.7-fold, 6.2-fold and 4.9-fold, respectively; Supplementary Fig. 3 and Table 3). Paneth cell markers were also downregulated in EED chips compared with healthy chips in control medium (for example, MT2A, CFTR and MT1H were suppressed by ~2–2.5-fold, respectively), but were differentially regulated when chips were exposed to nutritional deficiency. Healthy chips exposed to nutritional deficiency upregulated Paneth cell markers (for example, MT1H, MT1M and MT1G all increased by ~2-fold), while EED chips exposed to nutritional deficiency exhibited further Paneth cell marker downregulation (specifically, ID1, SULT1E1 and MT1X were reduced ~2–2.5-fold). These findings are consistent with the histopathological finding of Paneth cell depletion in EED patient intestinal biopsies38.
Intestinal villus atrophy and barrier compromise
As the transcriptomic analysis revealed downregulation in pathways involved in cell growth and intestinal barrier formation, we carried out differential interference contrast (DIC) and immunofluorescence microscopic analyses, which indeed confirmed that both healthy and EED intestine chips showed dramatically reduced growth of villus-like structures when cultured under nutrition deficient (−N/−T) conditions compared with healthy and EED control chips perfused with complete medium (Fig. 2a,b). Quantification of the height of the epithelium revealed that removal of these nutrients resulted in significant villus blunting in both healthy and EED chips in response to −N/−T deficiency, as indicated by a 70% and 80% reduction in epithelial height, respectively, compared with the same chips cultured in complete medium (Fig. 2c).
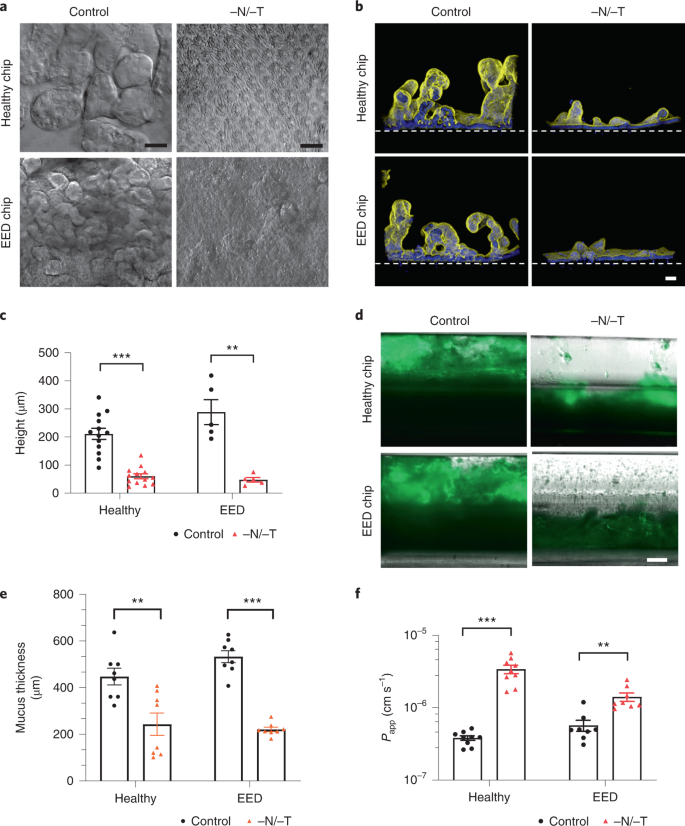
a, DIC imaging of intestine chips, top-down view (scale bar, 50 µm). b, Immunofluorescence cross-section micrographs showing villus-like structures in the intestine chips. Yellow, phalloidin; Blue, Hoechst; Dashed white line, upper surface of chip membrane. Scale bar, 50 µm. c, Villus-like structures height differences between control and −N/−T, healthy (mean: 211 µm for control and 60 µm for −N/−T) and EED (mean: 288 µm for control and 48 µm for −N/−T) intestine chips. Healthy, P < 0.000001, EED, P = 0.000685, multiple Student’s t-test. Each datapoint on the graph represents the average of 4 to 5 measurement points (membrane to top of the villi) measured in 2 to 3 cross-sectional images of the chip in at least 2 chips per condition. Two different healthy donors and one EED donor were used. Each chip corresponds to one biological replicate. d, Immunofluorescence microscopic views of longitudinal section through the two-channel intestine chips showing the mucus layer overlying the epithelium in the upper channel stained with Alexa 488-conjugated lectin. Scale bar, 250 µm. e, Graph showing differences in mucus thickness between control and −N/−T, healthy (mean: 448 µm for control and 243 µm for −N/−T) and EED (mean: 533 µm for control and 221 µm for −N/−T) intestine chips. Each datapoint on the graph represents the average of 2 measurement points measured in views similar to those shown in d; 2 chips per condition with chips created from cells from two different healthy donors and one EED donor. Healthy, P = 0.004092; EED, P < 0.000001, multiple Student’s t-test. Each chip corresponds to one biological replicate. f, Graph showing differences in Papp between control and −N/−T, healthy (mean: 3.82 × 10−7 for control and 3.41 × 10−6 for −N/−T) and EED (mean: 5.71 × 10−7 for control and 1.41 × 10−6 for −N/−T) intestine chips (9 for healthy chips and 8 for EED intestine chips). Healthy, P = 0.000005; EED, P = 0.001382, multiple Student’s t-test. Each chip corresponds to one biological replicate. Graphed data in c, e and f are mean ± s.e.m.
Source data
Another gene cluster identified as being preferentially sensitive to nutritional deficiency includes genes governing brush border structural integrity. These genes included myosin 1a (MYO1A), which links actin to the overlying apical membrane and whose absence results in irregularities of microvilli packing and length46; protocadherin-24 (CDHR2) that forms links between adjacent microvilli and is the target of enterohemorrhagic Escherichia coli-mediated brush border damage47; and mucin-like protocadherin (CDHR5) that forms heterophilic complexes with CDHR2 (Supplementary Fig. 4). Indeed, scanning electron microscopic imaging revealed that culturing healthy intestinal epithelium in −N/−T medium on-chip resulted in severe loss of apical microvilli (as well as links between adjacent microvilli) relative to control enterocytes that had their entire surface covered with tightly packed microvilli (Supplementary Fig. 5). Analysis of mucus accumulation by live imaging and staining with fluorescent lectin also revealed that both the healthy and EED chips exhibited a much thinner mucus layer when exposed to nutrient-deficient conditions (Fig. 2d,e).
Following these observations of structural changes due to exposure to nutritional deficiency, we next leveraged the advantage of using a two-channel microfluidic intestine chip (Fig. 1a) (that is, as opposed to intestinal organoids cultured within a solid extracellular matrix gel) to assess the effect of exposure to −N/−T medium on differences in intestinal barrier function between healthy and EED chips. We compared apparent permeability (Papp) values, which were measured by calculating the transfer of Cascade Blue fluorescent tracer (596 Da) from the epithelial lumen in the top channel to the underlying parallel channel below. Both healthy and EED control chips exhibited a tight barrier under baseline conditions (within the 10−7 Papp range) and displayed small but statistically significant reductions in barrier function when exposed to nutritional deficiency, as indicated by 8.9- and 2.5-fold increases in Papp for the healthy and EED chips, respectively (Fig. 2f).
Reduced nutrient absorption
Our transcriptomic analysis also revealed that nutritional deficiency resulted in the downregulation of multiple genes associated with the uptake and processing of important nutritional components, including fatty acids, certain amino acids and carbohydrates (Figs. 1e, 3a, 4a and Extended Data Fig. 2). This is clinically relevant because reduced absorption of nutrients is another hallmark of EED, and it affects weight and linear growth as well as cognitive development in children48,49,50,51. For example, expression levels for fatty acid translocase, cluster of differentiation 36 (CD36), microsomal triglyceride transfer protein (MTTP), apolipoprotein B (ApoB) and apolipoprotein C-III (ApoC3) were all lower in nutritionally deficient epithelium (Fig. 3a). Similarly, when we used immunofluorescence microscopy to assess expression of ApoB protein, which is a marker of chylomicron and fat metabolism in the intestine52, we found that exposure to −N/−T nutritional deficiency resulted in significant downregulation of ApoB in EED intestine chips (Fig. 3b and Supplementary Fig. 6). Furthermore, when we quantified cellular uptake of fatty acids using fluorescently labelled dodecanoic acid, we found that exposure to nutritional deficiency reduced fatty acid uptake by 1.68- and 1.69-fold in healthy −N/−T chips and EED −N/−T chips, respectively, compared with healthy and EED control chips (Fig. 3c). These findings are consistent with clinical data that similarly show impaired fatty acid metabolism in children suffering from EED16 and suggest that nutritional deficiency alone is sufficient to reduce fatty acid uptake even in healthy intestine.
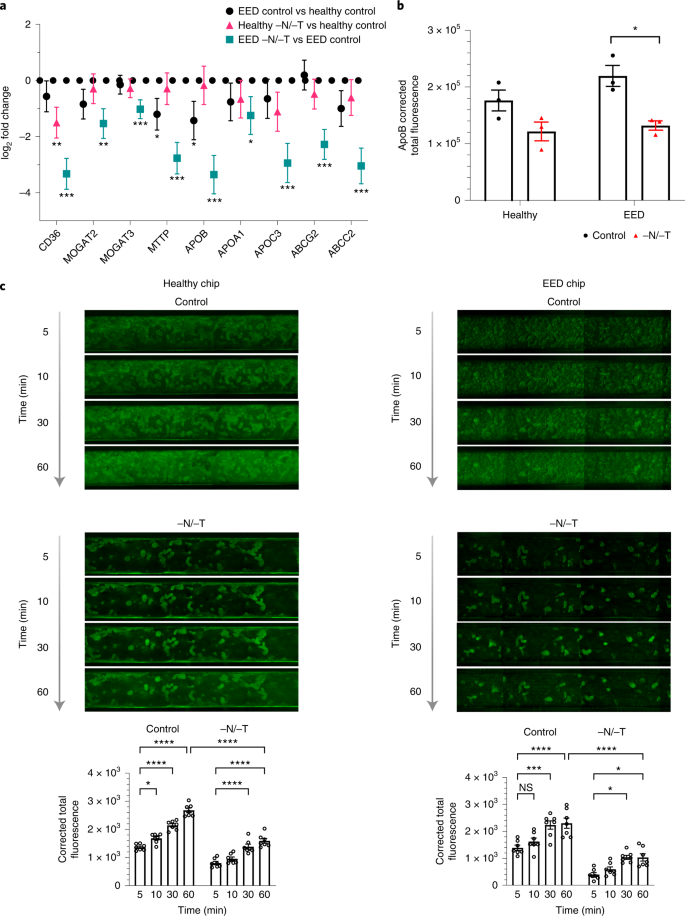
a, Transcriptional pathway analysis revealed a strong theme of downregulation for genes related to fatty acid uptake when EED chips were exposed to −N/−T media. The log2 fold changes between the average expression levels are depicted along with the associated 95% confidence interval. These changes included a 9.8-fold downregulation of the receptor CD36 (q < 0.001), a 10.2-fold downregulation of ApoB (q < .001) and an 8.2-fold downregulation of ABCC2 (q < 0.001). There was a similar trend towards downregulation when healthy chips were exposed to −N/−T media, but most gene changes did not reach statistical significance; 3 chips for each condition. Unmoderated Student’s t-test and the FDR method for multiple testing correction. Each chip corresponds to one biological replicate. b, Differences in ApoB corrected total fluorescence expression in control and −N/−T healthy and EED intestine chips. Healthy (mean: 1.7 × 105 for control and 1.2 × 105 for −N/−T), not significant; EED (mean: 2.2 × 105 for control and 1.3 × 105 for −N/−T), P = 0.012284. Each datapoint on the graph represents the average of corrected total fluorescence expression from 3 different measurement areas of intestine chip cross-sections, from at least 2 chips per condition. Each chip corresponds to one biological replicate. c, Differences in fluorescently labelled dodecanoic fatty acid uptake by healthy (left) and EED (right) intestine chips at the 5 min, 10 min, 30 min and 60 min timepoints. For each timepoint, 7 images covering the entire area of a representative chip from each condition were used to calculate the corrected total fluorescence expression (bottom). Graphed data in b and c are mean ± s.e.m. *P < 0.04, ***P ≤ 0.0008, ****P < 0.0001 by a 2-way ANOVA.
Source data
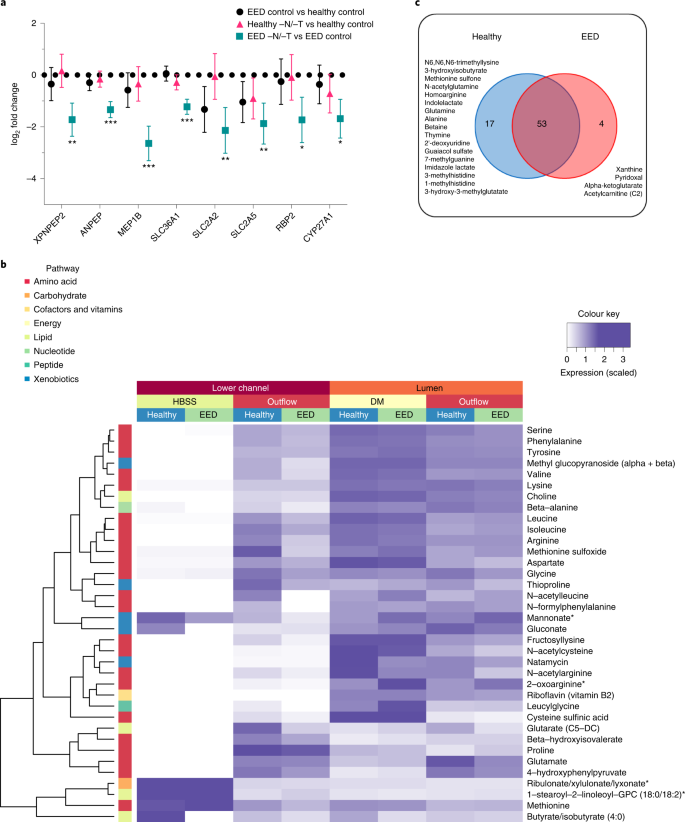
a, Amino acid processing and transport was among the strongly downregulated themes when EED chips were exposed to −N/−T media. This included a 6.2-fold downregulation of MEP1B (q < 0.001), a 4.4-fold downregulation of SLC2A2 (q = 0.0074) and a 3.3-fold downregulation of XPNPEP2 (q = 0.0036), which are shown as the log2 fold changes between the average expression levels along with the associated 95% confidence interval. There was occasional downregulation of these genes when healthy chips were exposed to −N/−T media, but none that achieved statistical significance; 3 chips for each condition. Each chip corresponds to one biological replicate. b, Heat map showing 36 metabolites taken up and transferred from the luminal medium to the lower channel of the intestine chips at higher abundance in the healthy vs EED chips (DM, differentiation medium; 3 chips for each condition). Each chip corresponds to one biological replicate. c, Venn diagram showing the number of common and unique metabolites secreted by the cells in intestine chips.
Source data
Abnormal amino acid uptake and metabolism
Children suffering from EED exhibit impaired development, and protein availability from the diet is a key factor responsible for linear growth; thus, we next explored differences in uptake by healthy and EED intestine chips. In the intestine, dietary protein is broken down into short peptides and free amino acids that are taken up by enterocytes, these short peptides and free amino acids serving as building blocks and energy sources for various organs and tissues48. Transcriptomic analysis revealed absorption and metabolizing factors that were downregulated in EED control chips vs healthy control chips, including the solute carrier family 2 (facilitated glucose transporter) member 2 (SLC2A2) and solute carrier family 2 (facilitated glucose/fructose transporter) member 5 (SLC2A5). Other nutrient transporters (such as amino acid absorption and metabolizing factors) that were found to be significantly downregulated in EED chips in response to −N/−T deficiency (EED −N/−T vs EED control), include SLC36A1, which encodes the proton-coupled amino acid transporter 1, ANPEP membrane enzyme alanyl aminopeptidase that is responsible for peptide digestion at the brush border, retinol binding protein 2 (RBP2), cytochrome P450, family 27, subfamily A, and polypeptide 1 (CYP27A1) (Fig. 4a and Supplementary Fig. 7).
We then assessed differences in absorption of nutrients between the healthy and EED intestine chips grown in control medium by performing untargeted metabolomic analysis by liquid chromatography tandem mass spectrometry (LC–MS/MS) to analyse epithelial uptake of nutrients and transfer of these molecules from the lumen of the intestinal epithelium in the top channel to the underlying basal channel (Fig. 1a). We detected 36 metabolites (out of >500 identified) that exhibited lower transport in EED intestine chips compared with healthy chips grown in control medium. These included mainly amino acid metabolites, but also metabolites related to nucleotides, cofactors, lipids, carbohydrates and xenobiotic pathways (Fig. 4b; an extended list of metabolites analysed can be found in Supplementary Fig. 8). Interestingly, 9 of these metabolites were amino acids previously identified as being reduced in serum of Malawian stunted children53. These included essential amino acids (isoleucine, leucine, methionine, phenylalanine, lysine), conditionally essential amino acids (arginine, glycine) and non-essential amino acids (glutamate, serine).
Our LC–MS/MS analysis also revealed 74 metabolites that were secreted by the intestinal cells as they were absent or at extremely low levels (<5%) in the perfusion medium. Of these metabolites, 17 were unique to the healthy control chips and included products of pathways related to metabolism of amino acids (N6,N6,N6-trimethyllysine, 3-hydroxyisobutyrate, methionine sulfone, N-acetylglutamine, homoarginine, indolelactate, glutamine, alanine, betaine, imidazole lactate, 3-methylhistidine, 1-methylhistidine), nucleotides (thymine, 2’-deoxyuridine, 7-methylguanine), xenobiotic metabolism (guaiacol sulfate) or lipids (3-hydroxy-3-methylglutarate). Interestingly, we also identified 4 metabolites unique to the EED chips cultured in control medium, including products of purine nucleotide metabolism (xanthine), vitamin B6 metabolism (pyridoxal), citric acid cycle/energy metabolism (alpha-ketoglutarate) and fatty acid metabolism (acetylcarnitine (C2)) (Fig. 4c).
To assess whether the observed differential uptake and metabolism of molecules in the intestine chips were transporter dependent, we quantified uptake of the dipeptide, glycyl-sarcosine (Gly-Sar), by the epithelial cells in the top channel, and its transfer to the lower channel using LC–MS/MS. This analysis revealed reduced uptake and transport of Gly-Sar in nutritionally deficient healthy intestine chips compared with the same chips cultured in control medium (Supplementary Fig. 9). In addition, these studies confirmed that these effects were due to transport through the PEPT1 transporter, and not due to passive inter- or intracellular diffusion, as this response could be completely prevented by adding Gly-Gly dipeptide (1 mM), which is a specific inhibitor of this transporter54 (Supplementary Fig. 9).
Altered inflammatory mediators
Altered intestinal inflammation is a key component of EED and our transcriptional analysis revealed that genes encoding key inflammatory marker proteins, such as lipocalin 2 (LCN2) and regenerating islet-derived protein 3 alpha (REG3A), were upregulated when EED or healthy chips were exposed to nutritional deficiency (Fig. 1c and Extended Data Fig. 3). Indeed, when we quantified the expression of nine key intestinal cytokines using a bead-based multiplexed ELISA assay, we detected higher levels of several cytokines (IL-6, ICAM, VCAM, IL-33, MCP-1, MIP-1 alpha and IL-8) in the epithelial lumen of healthy −N/−T intestine chips compared with the same chips cultured in control medium (Fig. 5a). The antimicrobial peptide Reg3A was also downregulated by nutritional deficiency in these chips. Interestingly, EED intestine chips grown in complete medium displayed reduced levels of all of the secreted cytokines analysed, while their levels were significantly upregulated when these chips were grown in nutritionally deficient medium (Fig. 5a). Similar responses were observed when we analysed cytokine levels in the lower parenchymal or vascular channel; however, significantly higher levels of the inflammatory cytokines IL-8, IP-10 and MCP-1 were observed (Fig. 5a). Interestingly, we found that intestine chips that were not exposed to mechanical peristalsis-like deformation secreted lower levels of IL-8 and MCP-1 (Extended Data Fig. 4).
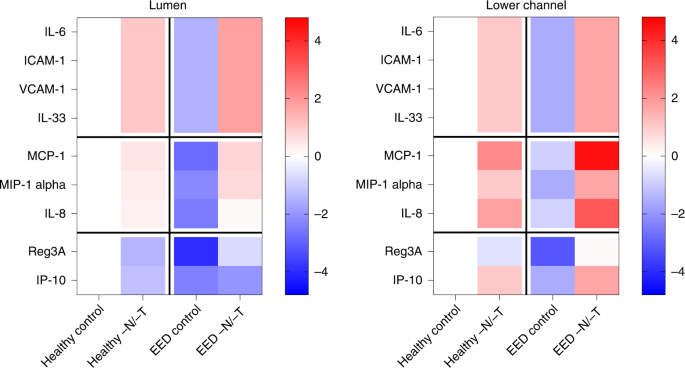
Heat maps showing differential expression of 9 cytokines secreted into the lumen or lower channel of the control or −N/−T, healthy and EED intestine chips and quantified using a bead-based multiplexed ELISA. The colour-coded scale represents the log2 fold change in expression; 3–6 chips for each condition. Each chip corresponds to one biological replicate.
Source data